The Food and Drug Administration (FDA or the Agency) plays a critical role in protecting the United States from threats such as emerging infectious diseases, including the Coronavirus Disease 2019 (COVID-19) pandemic. FDA is committed to providing timely guidance to support response efforts to this pandemic.
FDA is issuing this guidance to provide a policy to help expand the availability of surgical apparel for health care professionals, including gowns (togas), hoods, and surgeon’s and patient examination gloves during this pandemic.
This policy is intended to remain in effect only for the duration of the public health emergency related to COVID-19 declared by the Department of Health and Human Services (HHS), including any renewals made by the HHS Secretary in accordance with section 319(a)(2) of the Public Health Services (PHS) Act.
Medical gloves are used by health care personnel to prevent the spread of infection or illness. Medical gloves are disposable and include patient examination gloves and surgeon’s gloves.
To help expand the availability of medical gloves, the FDA is providing regulatory flexibility, as described in the enforcement policy for gloves that is in effect during the COVID-19 public health emergency. The FDA is also providing regular updates about medical gloves, including the answers to frequently asked questions on this page.
Standard for certification of FDA gloves
Go to the FDA official website and enter keywords to query the requirements of product certification FDA, such as querying gloves.
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm
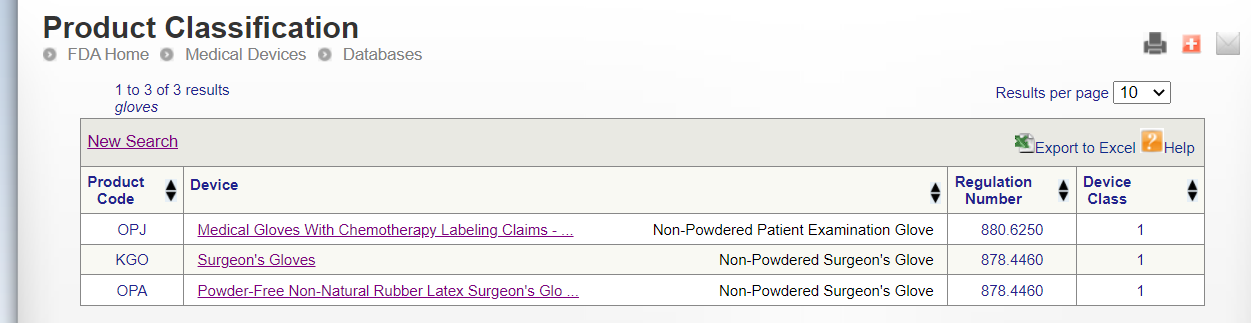
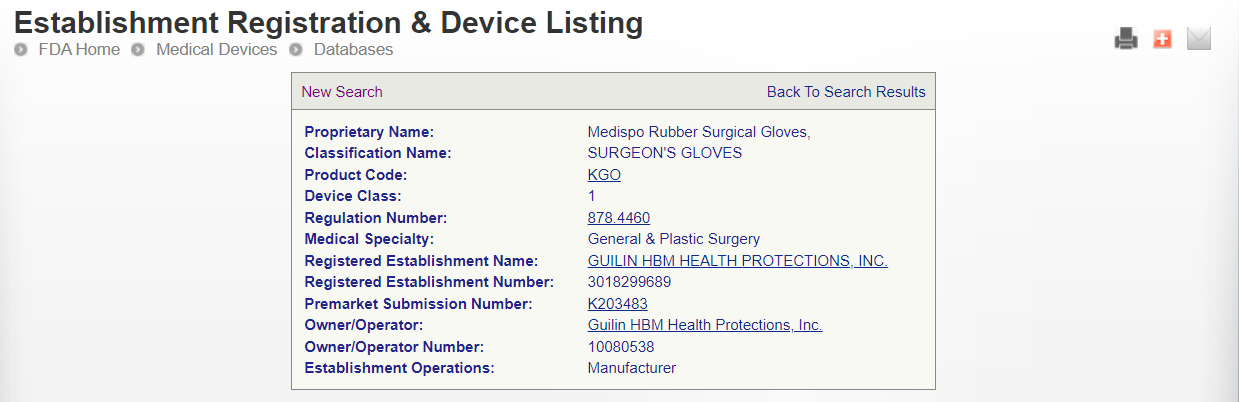
Surgeon’s Gloves FDA
Device: Surgeon’s Gloves
Regulation Description Non-powdered surgeon’s glove.
Definition
A surgeon’s glove is a disposable device made of natural rubber latex that may or may not bear powder to facilitate donning, and it is intended to be worn on the hands, usually in surgical settings, to provide a barrier against potentially infectious materials and other contaminants.
Regulation Medical Specialty: General & Plastic Surgery
Review Panel General Hospital
Product Code KGO
Premarket Review Surgical and Infection Control Devices (OHT4)
Infection Control and Plastic Surgery Devices (DHT4B)
Submission Type 510(k)
Regulation Number 878.4460
Device Class 1
Total Product Life Cycle (TPLC) TPLC Product Code Report
GMP Exempt? No
Summary Malfunction
Reporting Eligible
Implanted Device? No
Life-Sustain/Support Device? No
Medical Gloves With Chemotherapy Labeling Claims – Test For Use With Chemotherapy Drugs FDA
Regulation Description: Non-powdered patient examination glove.
Definition:
A disposable medical glove (examination or surgeons) that is worn on the examiner’s hand to prevent contamination between patient and examiner. In addition, these gloves have been tested for permeation and breakthrough resistance against various concentrations and types of chemotherapy drugs using test such as astm d6978-05, standard practice for assessment of resistance of medical gloves to permeation by chemotherapy drugs.
Physical State A disposable medical glove (examination or surgeons) that is worn on the examiner’s hand to prevent contamination between patient and examiner. In addition, these gloves have been tested for permeation and breakthrough resistance against various concentrations and types of chemotherapy drugs using test such as ASTM D6978-05, Standard Practice for Assessment of Resistance of medical Gloves to Permeation by Chemotherapy Drugs.
Technical Method A disposable medical glove (examination or surgeons) that is worn on the examiner’s hand to prevent contamination between patient and examiner. In addition, these gloves have been tested for permeation and breakthrough resistance against various concentrations and types of chemotherapy drugs using test such as ASTM D6978-05, Standard Practice for Assessment of Resistance of medical Gloves to Permeation by Chemotherapy Drugs.
Target Area:Glove
Regulation Medical Specialty: General Hospital
Review Panel: General Hospital
Product Code: OPJ
Premarket Review: Surgical and Infection Control Devices (OHT4)
Infection Control and Plastic Surgery Devices (DHT4B)
Submission Type 510(k)
Regulation Number: 880.6250
Device Class 1
Total Product Life Cycle (TPLC): TPLC Product Code Report
GMP Exempt? No
Summary Malfunction
Reporting Eligible
Implanted Device? No
Life-Sustain/Support Device? No
Powder-Free Non-Natural Rubber Latex Surgeon’s Gloves FDA Certification Requirements
Regulation Description: Non-powdered surgeon’s glove.
Definition
A powder-free surgeon’s glove is a disposable device made of non-natural rubber that may bear a trace amount of residual powder and it is intended to be worn on the hands, usually in surgical settings, to provide a barrier against potentially infectious materials and other contaminants.
Physical State A powder-free surgeon’s glove is a disposable device made of non-natural rubber that may bear a trace amount of residual powder and it is intended to be worn on the hands, usually in surgical settings, to provide a barrier against potentially infectious materials and other contaminants.
Technical Method A powder-free surgeon’s glove is a disposable device made of non-natural rubber that may bear a trace amount of residual powder and it is intended to be worn on the hands, usually in surgical settings, to provide a barrier against potentially infectious materials and other contaminants.
Target Area:Glove
Regulation Medical Specialty General & Plastic Surgery
Review Panel General Hospital
Product Code OPA
Premarket Review Surgical and Infection Control Devices (OHT4)
Infection Control and Plastic Surgery Devices (DHT4B)
Submission Type 510(k)
Regulation Number 878.4460
Device Class 1
Total Product Life Cycle (TPLC) TPLC Product Code Report
GMP Exempt? No
Summary Malfunction
Reporting Eligible
Implanted Device? No
Life-Sustain/Support Device? No
510(k) Third Party Review Program
The 510(k) Third Party Review Program provides medical device manufacturers with a voluntary alternative review process, in which accredited Third Party Review Organizations (3P510k Review Organizations) are allowed to review certain low-to-moderate risk medical devices. The program is intended to help yield more rapid 510(k) decisions and to allow the FDA to focus its resources on higher risk devices, while still maintaining oversight of the review of lower risk devices eligible for third party review. This program is formally known as the Accredited Persons Program.

Under the Third Party Review Program, a 510(k) submission for an eligible device may first be submitted to an accredited 3P510k Review Organization rather than directly to the FDA. Use of this program is voluntary. Approximately half of 510(k)s the FDA receives are eligible for this program. The sole payment under the program is between the 510(k) submitter and the 3P510k Review Organization; there is no separate payment (i.e., user fee) to the FDA.
3P510k Review Organizations use the same criteria used by the FDA to review 510(k) submissions. A 3P510k Review Organization’s review may include early interaction with the FDA to ensure the 3P510k Review Organization is using up-to-date standards and guidance relevant to that type of device. It may also include requests for additional information from the 510(k) submitter. After the 3P510k Review Organization is satisfied with its review and has documented all the necessary information for the submission, it sends the submission to the FDA including the original 510(k) submission, the 3P510k Review Organization’s review, and a recommendation of either substantially equivalent (SE) or not substantially equivalent (NSE).
The FDA makes the final determination on the 510(k) submission based on the review and recommendation received from the 3P510k Review Organization. If the 3P510k Review Organization did not appropriately apply the 510(k) decision criteria or if there are substantive review quality issues with their documentation, the FDA may need to re-review all or part of the 510(k) submission. However, the FDA is updating this program to avoid the routine re-review of 510(k) submissions already reviewed by a 3P510k Review Organization.
Learn how the FDA is eliminating the routine re-review of 510(k) submissions.
The FDA’s review timeframe for a MDUFA decision is within 30 days after receiving the recommendation of a 3P510k Review Organization.
There are four basic steps to submit a 510(k) through the program.
- Determine if your device is eligible.
To determine if a 510(k) submission is eligible to be reviewed under the Third Party Review Program, the 510(k) submitter can do any of the following:
- Review the list of eligible devicesfor the device product code (most Class I and Class II devices are eligible);
- Search the FDA’s Device Classification Database;
- Contact a Third Party Review Organization3P510k (Review Organization); or
- Contact the FDA at 3P510k@fda.hhs.gov.
- Find and contact a 3P510k Review Organization that can review your 510(k).
A 510 (k) submitter can use either of the methods below to find and contact organizations that can review their 510(k) submissions:
- Access the List of Devices for Third Party Review pageand then:
- Click on a device type at the bottom of the page. This will display a table with regulation names and product codes; and
- Click on the product code of the device. This will display additional information for the product code, including a list of any 3P510k Review Organizations that are eligible to review that type of device.
- Review the list of 3P510k Review Organizations (also referred to as Accredited Persons). The list of 3P510k Review Organizations shows the devices each organization is accredited to review, and their contact information.
- Obtain price quotes from one or more 3P510k Review Organizations and make a contract for a review. The fee for a review is determined by the agreement between the 510(k) submitter and the Review Organization. The 510(k) submitter pays the fee directly to the 3P510k Review Organization. The FDA does not collect a user fee for third party submissions.
- Submit the 510(k) to the 3P510k Review Organization. The submission should include the following:
- A letter authorizing the 3P510k Review Organization to discuss the 510(k) with the FDA and to forward it to the FDA on the 510(k) submitter’s behalf. The letter should include:
- Name of the 3P510k Review Organization;
- Name and contact information of the person assigned to the review; and
- Device trade name.
- The complete 510(k) submission, including the supporting data, summaries and analysis in the format requested by the 3P510k Review Organization.
- A letter authorizing the 3P510k Review Organization to discuss the 510(k) with the FDA and to forward it to the FDA on the 510(k) submitter’s behalf. The letter should include:
Interacting Early with the FDA During a 510(k) Review
The Third Party Review Program guidance outlines situations where Third Party Review Organizations (3P510k Review Organizations) should contact the FDA before or during their review of a 510(k) submission. This allows the FDA to provide 3P510k Review Organizations with answers to regulatory questions regarding current FDA processes and policies. Answers obtained through this process are intended to help 3P510k Review Organizations produce FDA-equivalent 510(k) reviews and recommendations and may eliminate FDA re-review of a 510(k) submission.
* If these timeframes cannot be met by the FDA, the FDA intends to work with the 3P510(k) Review Organization to establish a reasonable timeframe to provide feedback.
Prior to contacting the FDA, the 3P510k Review Organization is expected to conduct a review of all available resources to address certain questions. Resources may include:
- Relevant guidance documents, including device-specific and horizontal guidances (for example, biocompatibility, software, sterility) found in the FDA’s guidance database;
- Regulatory requirements for certain class II devices, also known as special controls, that apply to the device type;
- Postmarket databases for recallsand adverse events;
- FDA communications such as press releaseson recalls, market withdrawals, safety reports, and safety communications;
- Premarket review information about the predicate device to which a 510(k) submitter is comparing its device, or other similar device(s), including Indications for Use Statements, 510(k) Summaries, Decision Summaries, and the FDA’s decision letters. This information can be accessed in the FDA’s 510(k) database; and
- If an applicantant wishes to utilize standards, the 3P510k Review Organization should review the FDA’s guidance on “Appropriate Use of Voluntary Consensus Standards in Premarket Submissions for Medical Devices“.
Questions submitted to FDA should be clear, specific, and relevant to the 510(k) submission. They should be geared toward helping answer whether the device under review is substantially equivalent to the identified predicate device. Questions that lead to productive interactions between the 3P510k Review Organization and the FDA staff typically share the following characteristics:
- Take into consideration and include references to applicable guidance document(s) and standard(s):
- For instance, a 3P510k Review Organization can request clarification of a guidance or standard to ensure its interpretation is correct;
- A 3P510k Review Organization provides an explanation of why it believes information provided by a 510(k) submitter does not align with guidance or standards and then seeks FDA feedback on such assessment;
- Take into consideration postmarket data such as recalls and MDRs and previous discussions (for example, unsuccessful marketing applications, pre-submissions, etc.) with the FDA;
- Use previous FDA interactions on a third party 510(k) submission from their 3P510k Review Organization for that product code as a reference or starting point to see if any review practices have changed (provide submission numbers or copies of previous e-mail interactions); and
- Provide data as supportive context for a specific proposal, if necessary;
- For instance, a question may include a summary of nonclinical or clinical study data to help the FDA staff develop feedback in response to a specific proposal.
- 3P510k Review Organizations should notsubmit data or test protocols for pre-review. Likewise, the FDA should not request data or test protocols for pre-review.
Sample Questions
Regulatory Strategy Questions
- Are there concerns with the eligibility of the proposed device? While the product code (procode) XXX is Third Party eligible, this modification may fall under factor YYY in the guidance that may make this ineligible for third party review. FDA, do you concur that this device given the proposed modifications, appears eligible for third party review?
Sterilization Questions
- The 510(k) Submitter has followed standard XXX to validate their sterilization process. Their sterilization challenge device does not meet the requirements outlined in that standard; therefore, we intend to ask for additional information. Does FDA agree with this approach?
Guidance Questions
- The following guidance document XXX for this device type recommends that we should look for YYY data to demonstrate substantiate equivalence. The 510(k) submitter has instead provided ZZZ data. We believe that ZZZ data are sufficient as an alternative to address the risks outlined in the guidance for reasons A, B, and C. Does FDA agree with this?
To submit questions, the 3P510k Review Organization may call their FDA point of contact or send an email to 3P510k@fda.hhs.gov.
510(k) Third Party Performance Metrics and Accreditation Status
The Accredited Persons Program was created by the FDA Modernization Act of 1997 (FDAMA) to improve the efficiency and timeliness of the FDA’s 510(k) process. Under the program, the FDA accredits third parties (or accredited persons, now known as third party review organizations) that are authorized to conduct the primary review of 510(k)s for eligible devices. Under MDUFA IV, the FDA committed to publishing the performance of individual accredited Third Parties with at least five completed submissions on this page (for example, rate of not substantially equivalent (NSE), average number of holds, average time to substantially equivalent (SE)).
Accreditation Status
A list of accredited third party review organizations is Current List of FDA-Recognized 510(k) Third Party Review Organizations page.
Under section 523 of the Federal Food, Drug, and Cosmetic Act, the FDA may suspend or withdraw accreditation of any third party review organization, after providing notice and an opportunity for an informal hearing, when the accredited third party is substantially not in compliance with the requirements of the section, or poses a threat to public health, or fails to act in a manner that is consistent with the purposes of the section.
The FDA has withdrawn the recognition of third party review organizations listed in the table below:

How to check whether Chinese suppliers really pass FDA?
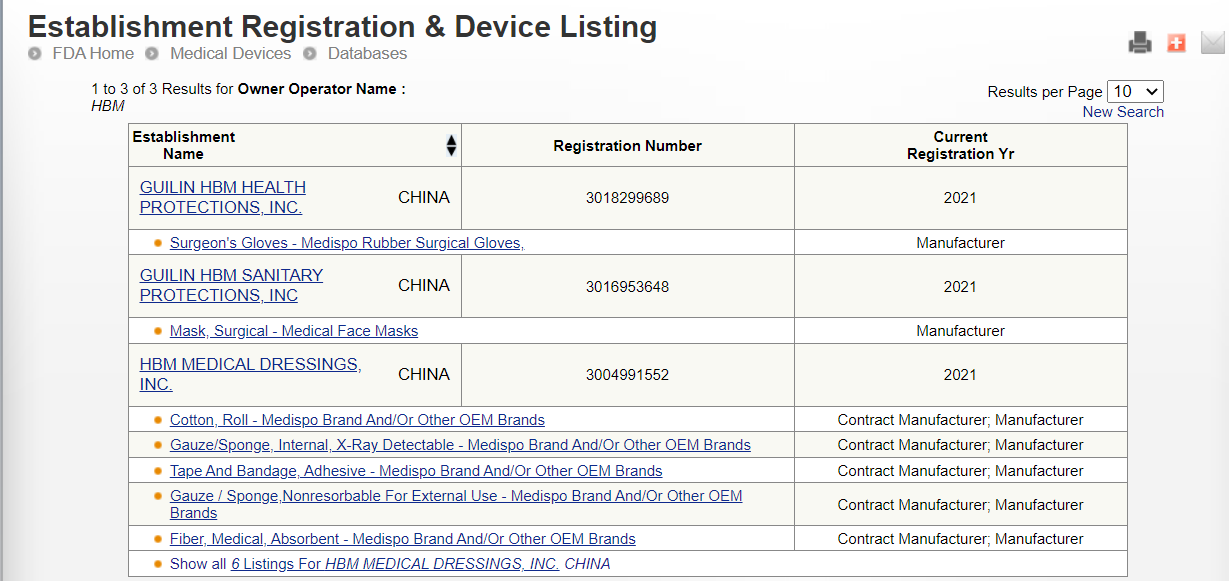
What does the FDA certificate look like?